- Forschung und Praxis
Grossgefässvaskulitiden: Riesenzellarteriitis und Takayasu-Arteriitis – Gemeinsamkeiten und Unterschiede
Die Riesenzellarteriitis und die Takayasu-Arteriitis gehören zu den Grossgefässvaskulitiden. Während erstere meist bei Menschen über 50 zu finden ist, beginnt letztere oft schon in der Kindheit oder Jugend. Ein Überblick.
15.10.2024
Zusammenfassung
Gemäss der Chapel-Hill-Konsensus-Konferenz-Nomenklatur von 2012 zählen die Riesenzellarteriitis (RZA) und die Takayasu-Arteriitis (TAK) zu den idiopathischen Grossgefässvaskulitiden. Während die RZA eine typische Erkrankung des älteren Menschen ist, beginnt die TAK im Adoleszenten- und jungen Erwachsenenalter, teilweise schon in der Kindheit. Bei beiden Erkrankungen kommt es durch eine immunvermittelte granulomatöse Gefässwandentzündung der Aorta und ihrer Hauptäste zur Ausbildung von Stenosen, Verschlüssen und Aneurysmen. Die Minderdurchblutung nachgeschalteter Organe führt zu den typischen Beschwerden wie Visusstörungen, ischämischen Muskelschmerzen und Dysästhesien. Typischerweise kommt es neben den lokalen Veränderungen zu einer ausgeprägten Systemreaktion mit konstitutionellen Symptomen wie Temperaturerhöhung, Abgeschlagenheit und Gewichtsverlust. Wird eine RZA oder eine TAK vermutet, sollte diese stets mittels bildgebender Verfahren (Ultraschall, MRI oder PET-CT) und/oder Histologie bestätigt werden. Neben der Induktionstherapie mit Glukokortikoiden haben steroidsparende Immunsuppressiva wie Tocilizumab und Infliximab in den letzten Jahren zunehmend an Bedeutung gewonnen, sodass die EULAR 2018 ihre Empfehlungen zum Management der Grossgefässvaskulitiden aktualisierten.
Der Artikel ist ursprünglich in der «Therapeutischen Umschau» (2022), 79(5), 221–228 erschienen.
Epidemiologie und Gefässbefall
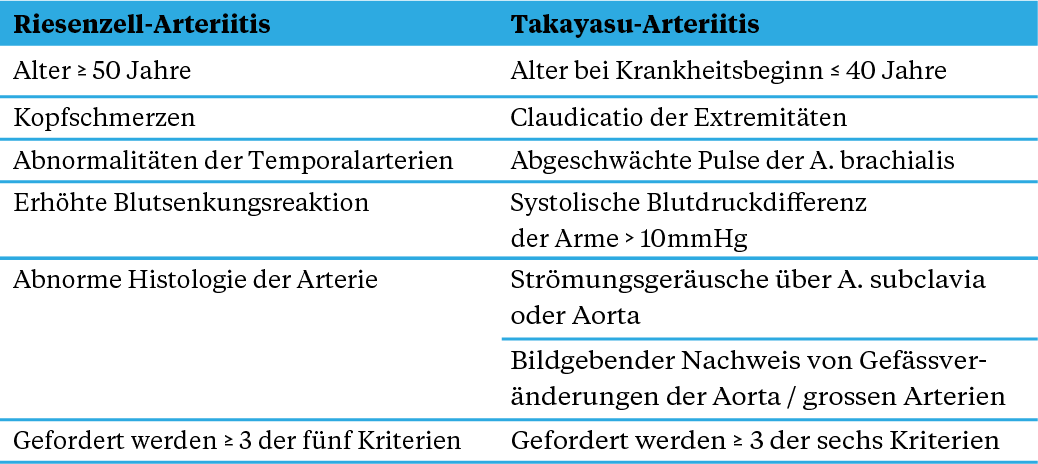
Gemäss der Chapel-Hill-Nomenklatur von 2012 werden die beiden idiopathischen Vaskulitisformen Riesenzellarteriitis (RZA) und Takayasu-Arteriitis (TAK) als Grossgefässvaskulitiden zusammengefasst [1]. Die RZA, auch Morbus Horton, Arteriitis temporalis oder kraniale Arteriitis genannt, ist die häufigste primäre Vaskulitisform des erwachsenen Menschen jenseits des 50. Lebensjahres. Weltweit beträgt die jährliche Inzidenz 1,6 bis 32,8 Fälle / 100 000 und erreicht typischerweise ihren Höhepunkt in der siebten bis achten Lebensdekade [2, 3]. 40 bis 60 Prozent der RZA-Patientinnen und -patienten weisen zusätzlich eine Polymyalgia rheumatica (PMR) auf. Obschon eine gleichzeitige RZA bei Vorliegen einer PMR seltener vorkommt (16 bis 29 Prozent), sollte diese bei Therapieresistenz, auch bei fehlender Klinik, gesucht werden [4, 5]. Die TAK ist mit einer jährlichen Inzidenz von 0,3 bis 2,2 Fälle / 100 000 weitaus seltener und beginnt meist in der Adoleszenz oder im frühen Erwachsenenalter [6, 7]. Eine TAK kommt in der Kindheit weniger häufig vor. Dennoch können in ca. einem Drittel der Fälle die ersten Symptome in die Kindheit rückdatiert werden [8]. Sowohl die TAK wie auch die RZA sind primär Erkrankungen des weiblichen Geschlechtes mit einem Frauen-zu-Männer-Verhältnis von ca. 9:1 bei der TAK und 3:1 bei der RZA [3, 7]. Lange Zeit galt die TAK typischerweise als Erkrankung asiatischer Frauen. Neuere epidemiologische Studien belegen jedoch, dass die TAK weltweit vorkommt und ein unterschiedliches Geschlechterverhältnis je nach Region aufweist [7]. Die RZA tritt ebenfalls weltweit auf, betrifft jedoch häufiger Personen kaukasischer Herkunft, insbesondere diejenigen mit skandinavischen Vorfahren [7]. In Tabelle 1 sind die «American College of Rheumatology (ACR)»-Klassifikationskriterien der RZA und der TAK von 1990 aufgelistet. Kürzlich wurden neue ACR-Klassifikationskriterien ausgearbeitet, die 2022 publiziert wurden. Wichtig ist, dass diese Kriterien primär für eine Klassifikation (zum Beispiel in Studien) und nicht als Diagnosekriterien erstellt wurden.
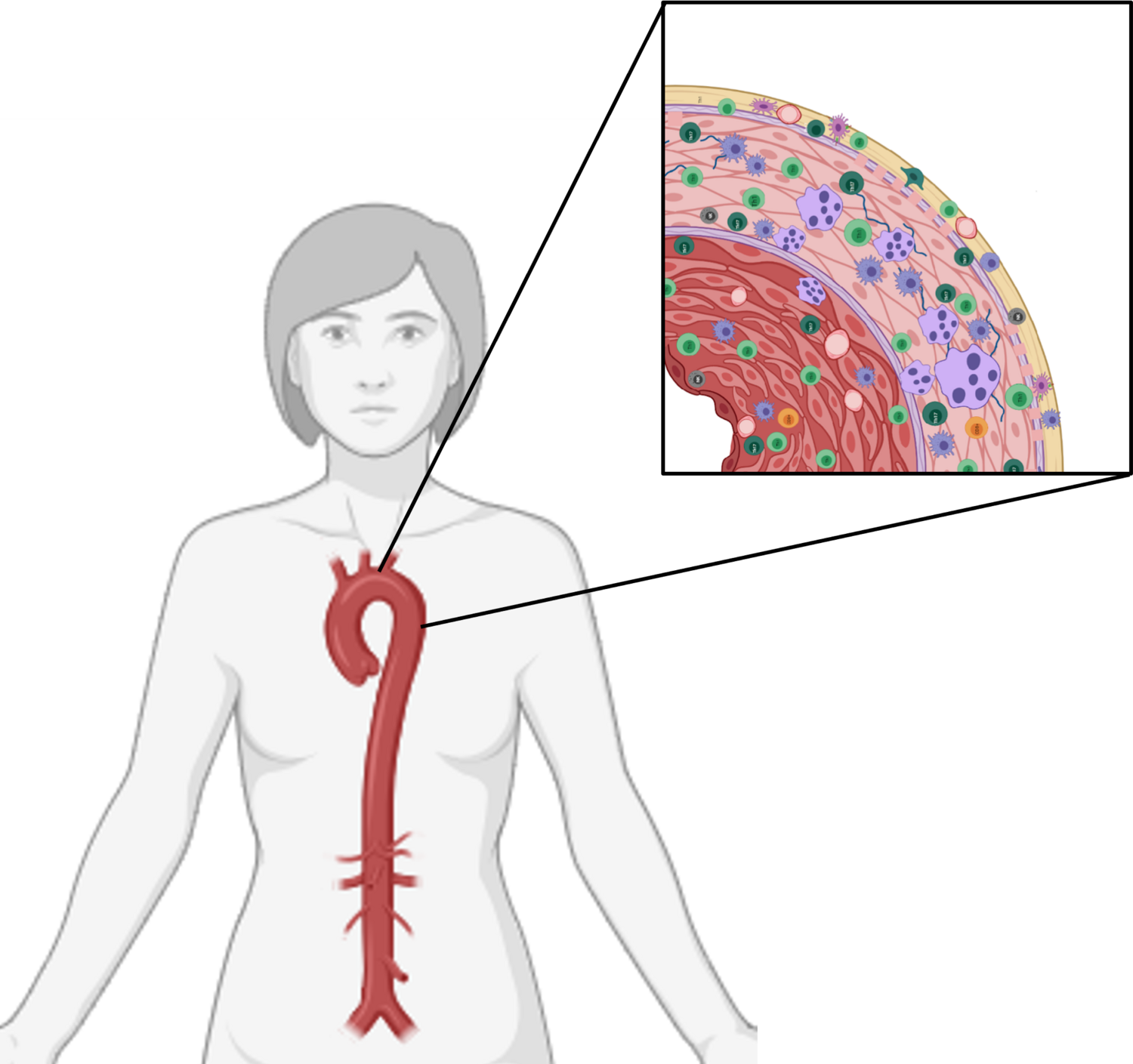
Während die TAK fast ausschliesslich die Aorta, deren Abgänge sowie die Karotiden betrifft, befällt die RZA tendenziell peripherer gelegene, mittelgrosse bis kleine Gefässe wie Temporalarterien und die A. Centralis retinae (Abb. 1). In etwa der Hälfte der RZA-Fälle kann eine prognostisch relevante Beteiligung der Aortenwand nachgewiesen werden und sollte daher stets gesucht werden [9, 10]. Trotz der vielen Ähnlichkeiten zwischen der RZA und der TAK liessen sich durch neuere bildgebende Verfahren sowie genetische und molekularbiologische Forschungsmethoden merkliche Unterschiede in der Pathophysiologie, im Gefässbefall und im Krankheitsverhalten nachweisen, was insbesondere für die Wahl der geeigneten Therapie sowie für das Krankheitsmanagement von entscheidender Bedeutung ist [11, 12].

Ätiologie und Pathogenese
Das unterschiedliche Alter sowie das geografische und geschlechtsspezifische Auftreten der RZA und der TAK deuten darauf hin, dass Alterungsprozesse, biologisches Geschlecht, genetischer Hintergrund und Umweltfaktoren zur Entstehung und Aufrechterhaltung der RZA und der TAK beitragen [3, 7].
Auflösung des Immunprivilegs – Einwanderung von T-Zellen und Makrophagen: Die arterielle Gefässwand ist aus den drei Schichten Intima, Media und Adventitia aufgebaut. Während kleine Arterien durch Diffusion aus dem Intravasalraum mit Sauerstoff und Nährstoffen versorgt werden, gewährleistet bei grösseren Arterien ein Netz aus kleinen Gefässen (Vasa vasorum) die Versorgung der Media und Adventitia. Die Vasa vasora werden im Bereich der Media-Adventitia-Grenze von vaskulären dendritischen Zellen (vasDC) umgeben. Unter physiologischen Bedingungen ist die Gefässwand der Arterien «immunpriviligiert», was bedeutet, dass ausser dieser vasDC keine Immunzellen in der Gefässwand vorkommen und somit eine spontane Immunreaktion gegen Selbstantigene verhindert wird. Bei der RZA und TAK führt ein unbekannter Auslöser zu einer pathologischen Aktivierung dieser vasDC, was zur Aufhebung des Immunprivilegs führt. Zirkulierende T-Zellen und Monozyten werden durch Chemokine wie CCL-19 und -21 (freigesetzt von vasDC) angelockt und wandern über die Vasa vasora in die Gefässwand ein [13, 14]. Normalerweise verhindert die Basalmembran der Vasa vasora das Eindringen von Immunzellen in die Gefässwand. Monozyten von RZA-Patientinnen und -Patienten produzieren jedoch erhöhte Mengen an Matrix-Metalloproteasen (MMP), insbesondere MMP-2 und MMP-9, die in der Lage sind, das Kollagen IV der Basalmembran abzubauen. Hierdurch wird es anderen Immunzellen (insbesondere T-Zellen) ermöglicht, in die Gefässwand einzudringen [15]. T-Zellen und Monozyten-basierte Makrophagen formieren sich zusammen zu Granulomen, Makrophagen fusionieren und bilden mehrkernige Riesenzellen. Hohe Mengen an entzündungsfördernden Zytokinen wie MMP, Interferon gamma (IFNγ), Tumor necrosis factor alpha (TNF-α), IL-1b, IL-2, IL-6, IL-17, IL-21 und IL-22, Wachstumsfaktoren und pro-angiogenetische Faktoren wie «platelet derived growth factor» (PDGF) und «vascular endothelial growth factor» (VEGF) werden freigesetzt. Es entsteht eine lokale Gefässwandentzündung mit Degeneration der Extrazellulärmatrix, Intimahyperplasie und Fibrosierung (Abb. 2) [13, 16]. vasDC exprimieren kostimulierende und koinhibierende Moleküle auf ihrer Oberfläche, durch die sie die Aktivität der umgebenden T-Zellen regulieren. Eines dieser koinhibierenden Signale wird durch die Interaktion zwischen dem «programmed cell death protein 1» (PD-1, exprimiert auf T-Zellen) und seinem Liganden PD-L1 (exprimiert auf vasDC) vermittelt. vasDC von RZA-Patientinnen und -Patienten exprimieren kaum PD-L1, wodurch diese nicht in der Lage sind, Effektor-T-Zellen zu blockieren. Es kommt zu einer lokalen Ansammlung und vermehrten Aktivierung dieser Effektor-T-Zellen. Im Gegensatz zur RZA ist deutlich weniger bekannt über die Rolle von vasDC und MMP bei der Entstehung der TAK. Das Entzündungsinfiltrat bei der TAK ähnelt jedoch stark dem der RZA, mit der typischen Ausbildung von Granulomen und mehrkernigen Riesenzellen. Wie auch bei der RZA finden sich vasDC in unmittelbarer Nähe zu T-Zellen, was vermuten lässt, dass diese ebenfalls an der T-Zell-Aktivierung beteiligt sind [17]. Ob MMP auf ähnliche Weise an der Pathogenese der TAK beteiligt sind, ist umstritten. Während in einigen Studien erhöhte Blut-MMP-Werte (MMP-2, -3, -9) bei TAK-Patienten gefunden wurden, konnten in anderen Studien keine Unterschiede im Vergleich zu gesunden Kontrollen festgestellt werden [18].

T-Zellen, natürliche Killerzellen: Sowohl bei der RZA wie auch bei der TAK spielen CD4+-T-Zellen, insbesondere INFγ-produzierende Th1- und IL-17-produzierende Th17-Zellen, eine entscheidende Rolle bei der Auslösung und Aufrechterhaltung der Gefässentzündung. Im Gegensatz zur RZA scheinen bei der TAK auch CD8+-T-Zellen und natürliche Killerzellen (NK) wesentlich zur Gefässzerstörung beizutragen. Immunhistochemische Färbungen des Gefässwandinfiltrates zeigen einen deutlich höheren Anteil an CD8+-T-Zellen und NK-Zellen bei der TAK im Vergleich zur RZA [19, 20]. Eine Erklärung für dieses unterschiedliche CD4+/CD8+-T-Zellen-Verhältnis liefert der Einfluss des Alterns auf die T-Zell-Kompartimente. Im Vergleich zu CD4+-T-Zellen kommt es bei CD8+-T-Zellen zu einem deutlich stärkeren Rückgang der relativen und absoluten Zellzahl und zu einer ausgeprägteren Expression von Alterungsmarkern mit zunehmendem Alter [21]. Ein anderer Faktor, der einen Einfluss auf die Zusammensetzung des Entzündungsinfiltrats haben könnte, liegt in den Genen. Als stärkster genetischer Risikofaktor für die Entwicklung einer RZA oder einer TAK konnten Polymorphismen in den Haupthistokompatibilitätskomplexen-I und -II (Englisch: «major histocompatibility complex» [MHC-I & MHC-II]) identifiziert werden. Über MHC-I-Proteine werden zelleigene Antigene mehrheitlich CD8+-T-Zellen und NK-Zellen präsentiert, während über MHC-II extrazelluläre, phagozytierte Antigene hauptsächlich CD4+-T-Zellen präsentiert werden [22]. Die RZA ist assoziiert mit Veränderungen in den HLA-Regionen («human leukocyte antigen») des MHC-II-Gens, während die TAK Polymorphismen im MHC-I-Gen aufweist [23]. Ein interessanter Unterschied zwischen der RZA und der TAK ist das unterschiedliche Therapieansprechen von CD4+-T-Zellen. Th17-Zellen von RZA-Patientinnen und -Patienten sprechen gut auf Glukokortikoide (GC) an, während Th1-Zellen trotz längerer Behandlung in den vaskulären Läsionen persistieren. Im Gegensatz dazu sind Th1-Zellen von TAK-Patientinnen und -Patienten empfindlich gegenüber GC, und Th17-Zellen sind steroidresistent [24, 25]. Die regulatorische T-Zelle (Treg) ist ein weiterer T-Zellsubtyp, der an der Pathogenese der RZA und der TAK beteiligt ist. Bei RZA-Patientinnen und -Patienten mit aktiver Erkrankung weisen Treg eine veränderte Expression des «Forkhead box proteins P3» (FoxP3) auf, wodurch diese ihre suppressive Funktion verlieren und durch die vermehrte Ausschüttung von IL-17 sogar krankheitsfördernd werden. Tocilizumab (TCZ), ein monoklonaler IL-6-Rezeptor-Antikörper zur Therapie der RZA, führt zu einer Normalisierung der Treg-Zellfunktion, verstärkt deren Suppressionskapazität und bewirkt eine Reduktion von IL-17 [26]. Ein spezieller Treg-Zell-Subtyp, die CD8+-Treg-Zelle, kontrolliert umliegende CD4+-T-Zellen durch die Freisetzung von enzymhaltigen Mikrovesikeln. Bei der RZA kommt es zu einer Störung des intrazellulären Transportes und der Freisetzung dieser Mikrovesikel. Diese verminderte CD4+-T-Zell-Inhibition führt zur Proliferation und Aktivierung von CD4+-T-Zellen [27]. Obwohl es Hinweise auf eine mögliche Dysfunktion der Treg-Zellen bei der TAK gibt, ist deutlich weniger über ihre Funktion und Beteiligung am Krankheitsgeschehen bekannt, und die Studienresultate sind teilweise widersprüchlich.
mTORC1-Hyperaktivität: Ein weiterer wichtiger Schlüsselmechanismus in der Pathogenese der RZA und der TAK ist der mTORC1(«mechanistic Target of Rapamycin Complex 1»)-Signalweg. mTORC1 ist ein entscheidender Regulator von Zellwachstum, Zellmetabolismus und Zelltod. Bei beiden Erkrankungen kommt es zu einer Hyperaktivierung von mTORC1, was zu einer Vermehrung von Endothelzellen, zur erhöhten Freisetzung von entzündungsfördernden Zytokinen, zur Förderung der CD4+-T-Zellproliferation in Richtung Th1- und Th17-Zellen und zur Beeinträchtigung der Treg-Zellfunktion führt [28]. Bei der RZA wird die vermehrte mTORC1-Aktivität durch zwei dysfunktionale Upstream-Mechanismen verursacht. Erhöhte VEGF-Werte führen zu einer verstärkten Expression des NOTCH1-Ligand Jagged1 auf Endothelzellen der Vasa vasora. Die Interaktion zwischen Jagged1+-Endothelzellen und NOTCH1+-T-Zellen, führt zur Aktivierung von mTORC1 in den T-Zellen [29]. mTORC1 wird ebenfalls durch eine vermehrte Aktivität des CD28-PI3K-AKT-Signalweges verursacht [30]. Bei der TAK wurden diese beiden Signale bis anhin nicht untersucht, jedoch konnten Hadjadj et al. zeigen, dass es durch die Bindung von IgG-Antikörpern an Endothelzellen zu einer vermehrten mTORC1-Aktivierung kommt [31]. Auch T-Zellen weisen bei der TAK eine vermehrte mTORC1-Aktivität auf, die verantwortlichen Mechanismen sind noch nicht untersucht [28].
Klinik
Die RZA kann sich in unterschiedlichen Phänotypen manifestieren, die einzeln oder überlappend vorkommen können. Zum einen gibt es die klassische kranielle RZA (Arteriitis temporalis) mit dem Befall der Arteriae carotides externa und deren Abgänge. Ein Befall dieser Gefässe führt zu den typischen Symptomen wie Schläfenkopfschmerz, Dysästhesie der Kopfhaut, Kauclaudicatio und Sehstörungen (wie Doppelbilder oder Visusverlust). Zum anderen kann die RZA mit Symptomen einer PMR assoziiert sein, die sich mit Muskelschmerzen und Steifheit im Hüft- und Schultergürtel präsentiert. Beim dritten Phänotyp kommt es zu einer Entzündungsreaktion und einem strukturellen Umbau der Gefässwand der Aorta und deren Abgänge, was im Verlauf zur Bildung von Stenosen und Aneurysmen führen kann. Häufig wird der lokale Prozess von einer ausgeprägten systemischen Entzündungsreaktion mit deutlicher B-Symptomatik, z. B. Gewichtsverlust, Abgeschlagenheit und Fieber, begleitet [32]. Eine gefürchtete Komplikation der RZA ist die Augenischämie mit irreversiblem Sehverlust [33, 34]. Eine Augenbeteiligung kommt bei 10 bis 30 Prozent der RZA-Patientinnen und -Patienten vor und manifestiert sich meist in Form einer anterioren ischämischen Optikusneuropathie. Weitere Manifestationen wie posteriore ischämische Optikusneuropathie oder kortikale Blindheit sind weitaus seltener. In bis zu 15 Prozent der Fälle kommt es zu einem irreversiblen Sehverlust. Eine schnellstmögliche Therapieeinleitung mit GC ist entscheidend, um einen persistierenden Sehverlust zu vermeiden, und sollte bei hohem Verdacht auf eine RZA bereits vor Diagnosesicherung erfolgen. Die Ausbildung eines Aortenaneurysmas mit der Gefahr einer Dissektion oder Ruptur tritt teilweise mit zeitlicher Latenz auf, weshalb ein Langzeitmonitoring entscheidend ist [34].
Die TAK ist gekennzeichnet durch einen chronisch schleichenden, subakuten Verlauf mit Phasen stärkerer und schwächerer Aktivität. Dies führt oft zu einer Diagnoseverzögerung. Die frühe Krankheitsphase ist geprägt durch unspezifische konstitutionelle Symptome, wie Abgeschlagenheit, Gewichtsverlust, leichte Temperaturerhöhung (meist weniger ausgeprägt als bei der RZA) und Nachtschweiss, und lässt kaum eine TAK vermuten. Mit zunehmender Krankheitsdauer kommt es durch die chronische Gefässwandentzündung zu Stenosen, Okklusionen und aneurysmatischen Erweiterungen der Aorta und ihrer Hauptäste, was zu den typischen Symptomen und Befunden und infolgedessen zur Diagnosefindung führt. Das Leitsymptom ist die ischämisch bedingte Armclaudicatio. Tritt diese zusammen mit den klassischen Befunden eines brachialen Pulsdefizits (typisch über der Arteria radialis), einer Blutdruckdifferenz zwischen rechtem und linkem Arm und eines Stenosegeräuschs über den grossen Arterien auf, ist eine TAK sehr wahrscheinlich [6]. Die häufigsten Symptome der RZA und TAK sind in Abbildung 1 zusammengefasst.
Diagnosestellung
Bei Diagnosestellung ist neben der Anamnese die Erhebung des klinischen Befunds inklusive Gefässstatus essenziell. Der Gefässstaus umfasst die Auskultation aller grossen arteriellen Gefässe des Rumpfes inklusive der A. axillaris und A. brachialis, ferner die Beurteilung des Pulses und die seitenvergleichende Blutdruckmessung, bei der TAK an allen vier Extremitäten. Bei der TAK kann hierbei in bis zu 90 Prozent der Fälle durch eine körperliche Untersuchung bereits klinisch eine Diagnose gestellt werden [6].
Im Blut sollte eine Messung der Entzündungsparameter (CRP, BSR) erfolgen. Zu beachten ist hierbei, dass eine fehlende Inflammation eine aktive RZA nicht ausschliesst. Weitere Blutparameter und Autoantikörper (wie ANCA) können in der Routine bei der TAK und der RZA zur Abgrenzung von Differenzialdiagnosen oder zur Erfassung von Komorbiditäten dienen.
Die Diagnose einer Grossgefässvaskulitis sollte immer mittels Histologie und/oder Bildgebung objektiviert werden. Bei RZA-Verdacht wird die Temporalarterien-Biopsie (TAB) heutzutage in der Regel einseitig durchgeführt (Präparatlänge optimal > 1 cm) und wenn möglich innert zwei Wochen nach Beginn der GC-Therapie [35]. Zu beachten gilt, dass wegen des segmentalen Befalls eine negative TAB eine RZA nicht ausschliesst. Eine Histologie zur Diagnosesicherung der TAK ist nicht üblich. Neben der Histologie stehen mittlerweile eine Reihe an bildgebenden Verfahren zur Verfügung, die einzeln oder in Kombination eingesetzt werden: FDG-PET / CT, MRT / MR-Angiographie der Aorta, CT / CT-Angiographie und Ultraschall (Abb. 3). Fast-Track-Clinics und der schnelle Zugang zu Bildgebungen, wie der Ultraschalldiagnostik, erlauben eine raschere Diagnostik, was sich in einer besseren Prognose widerspiegelt [36]. Die Wahl der Bildgebung erfolgt unter Berücksichtigung der lokal vorhandenen Ressourcen und Patientenfaktoren (wie Kontraindikation für MR). Wichtig ist, dass relevante Strukturen dargestellt werden. Hierzu zählen insbesondere die A. carotis, A. subclavia und Aorta bei der TAK und die kraniellen, aber auch die grossen Gefässe inklusive Aorta bei der RZA.
Eine Bildgebung der Aorta sollte, wenn immer möglich, bei allen RZA-Patientinnen und -Patienten bei Diagnosestellung und unter möglichst geringer GC-Exposition erfolgen. Bei der TAK sollte aufgrund des jüngeren Alters eine strahlenfreie Untersuchung wie die MRT bevorzugt werden. Die invasive konventionelle Angiographie wird heutzutage kaum noch zur Diagnosestellung eingesetzt. Aufgrund der meist langwierigen und potenziell nebenwirkungsreichen Therapie ist eine solide Diagnostik bei Diagnosestellung unverzichtbar.

Therapie
Erfreulicherweise hat sich das Therapiespektrum sowohl bei der RZA wie auch bei der TAK erweitert. Nebst GC, die bei längerer Therapiedauer fast unweigerlich mit Nebenwirkungen einhergehen, stehen GC-sparende Therapien wie TNF-α-Inhibitoren (TNFi) oder TCZ zur Verfügung. Therapieempfehlungen wurden unter anderem von der EULAR, der deutschen Gesellschaft für Rheumatologie (DGRh) und der ACR publiziert [35, 37, 38].
Eine akute RZA stellt einen medizinischen Notfall dar. Nebst einer umgehenden Evaluation durch eine spezialisierte Ärztin oder einen spezialisierten Arzt mit Zugang zu geeigneten diagnostischen Methoden soll die Therapie bei begründetem Verdacht auf eine RZA bereits vor Diagnosesicherung begonnen werden. Das Therapieziel ist das Erreichen einer Remission, die ohne vaskulitis-assoziierte Organschäden erreicht werden sollte. Die Definition einer Remission basiert auf einem Expertenkonsens und erfordert die Abwesenheit von Vaskulitis-Symptomen, eine Normalisierung der Entzündungsparameter und den Ausschluss von progredienten Gefässveränderungen [37].
Glukokortikoide
Obwohl effektiv, sollten die kumulative und zeitliche GC-Expositionsdauer wann immer möglich aufgrund des Nebenwirkungsprofils so tief wie möglich gehalten werden. Hochdosierte orale GC werden bei Diagnosestellung einer aktiven RZA oder TAK empfohlen (40–60 mg/d Prednisolon-Äquivalent). Bei TAK-Patientinnen und -Patienten ohne ischämische Symptome sollten tiefe GC-Startdosen rezeptiert werden. Eine intravenöse GC-Pulstherapie ist bei RZA-Patientinnen und -Patienten mit visuellen Symptomen (akuter Visusverlust oder Amaurosis fugax) sinnvoll [35, 37, 38]. Rational hierbei ist, theoretisch vom schneller einsetzenden, nicht genomischen Effekt einer GC-Pulstherapie zu profitieren [39].
Bei kontrollierter Krankheitsaktivität sollte ein Ausschleichen der GC erfolgen. Hierbei variieren die Empfehlungen hinsichtlich der Geschwindigkeit der GC-Dosisreduktion. Es empfiehlt sich im Alltag, einen vordefinierten Dosiswert nach zwölf Wochen Therapie anzustreben (zum Beispiel 10 bis 15 mg/d bei der RZA, 5 mg/d bei der TAK).
GC-sparende Therapien
In den meisten Fällen ist eine GC-sparende Therapie sinnvoll. Anhand der aktuellen Studienlage sind TNFi und TCZ bei der TAK gleichwertig, bei der RZA ist nur die Wirkung von TCZ belegt [35].
Bei der RZA konnten zwei randomisiert kontrollierte Studien (RCT) eine eindrückliche GC-sparende und remissionserhaltende Wirkung von TCZ zeigen [40, 41]. In der GUSTO-Studie konnte bei aktiver RZA eine Remission erzielt werden durch eine TCZ-Monotherapie nach vorangehender nur dreitägiger GC-Therapie [42]. Dies unterstreicht den Stellenwert von TCZ als GC-sparende Therapie bei der RZA. Unter TCZ-Therapie ist zu beachten, dass die CRP-Werte supprimiert sind und daher nicht zur Beurteilung der Krankheitsaktivität verwendet werden können.
Als Therapiealternative findet sich Methotrexat (MTX) (off-label), wobei Kontraindikationen zu berücksichtigen sind (wie eine schwere Niereninsuffizienz oder Leberfunktionsstörung). In einer Metanalyse aus drei RCT konnten für MTX eine tiefere Rückfallrate und kumulative GC-Exposition gezeigt werden [43]. Mit Abatacept (off-label, Phase-II-Daten vorliegend, Phase-III-Studie laufend) steht ein weiteres Biologikum zur Verfügung, das vor allem bei Kontraindikation und Insuffizienz von TCZ und MTX erwogen werden kann [35]. Zudem werden derzeit eine Fülle an weiteren Substanzen in klinischen Studien untersucht, wie Biologika (beispielsweise Anakinra, Secukinumab oder Mavrilimumab) oder Upadacitinib, ein JAK-Inhibitor [44].
TAK-Patientinnen und -Patienten weisen in bis zu 70 Prozent der Fälle einen rezidivierenden Verlauf auf, und die Dosisreduktion einer GC-Monotherapie gestaltet sich oft schwierig. Daher ist in der Regel immer eine GC-sparenden Therapie angezeigt. Hierbei kommen in der Regel zunächst csDMARD zum Einsatz wie MTX (Kontraindikation in der Schwangerschaft) oder Azathioprin [37]. Ein Kinderwunsch sollte bei der Therapieplanung berücksichtigt werden. Biologika werden oft in zweiter Linie eingesetzt. Hierbei sind TNFi einer TCZ-Therapie bei TAK-Patientinnen und -Patienten vorzuziehen [35].
Bei der TAK besteht ein gutes Potenzial zur Kollateralausbildung, weswegen primär ein medikamentöses Management angestrebt werden sollte. Falls ein interventioneller oder gefässchirurgischer Eingriff notwendig ist, sollte die Indikation hierfür interdisziplinär gestellt werden und der Eingriff, falls möglich, bei stabiler Remission erfolgen.
Ergänzend sind ein Screening für therapie-assoziierte und kardiovaskuläre Komorbiditäten sowie die entsprechenden Impfungen angezeigt.
Die Therapiedauer bei der RZA beträgt mindestens ein Jahr. Im Falle eines Rezidivs, ischämischer Komplikationen oder beim Vorliegen von Risikofaktoren (wie extrakranieller Befall) sollte eine Therapiedauer über ein Jahr hinaus erwogen werden. Die Verlaufskontrollen sollten nach individuellem Ermessen durchgeführt werden. In aller Regel empfiehlt sich im ersten Jahr eine engmaschigere Kontrolle alle ein bis drei Monate, die bei gutem Verlauf ab dem zweiten Jahr auf drei- bis sechsmonatliche Kontrollen ausgedehnt werden kann. Aufgrund der Möglichkeit von Rezidiven und Gefässkomplikationen im Langzeitverlauf empfiehlt sich eine Verlaufskontrolle über mehrere Jahre.
Literatur
- Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013;65(1):1–11.
- Lee JL, Naguwa SM, Cheema GS, Gershwin ME. The geo-epidemiology of temporal (giant cell) arteritis. Clin Rev Allergy Immunol. 2008;35(1–2):88–95.
- Berti A, Dejaco C. Update on the epidemiology, risk factors, and outcomes of systemic vasculitides. Best Pract Res Clin Rheumatol. 2018;32(2):271–94.
- Buttgereit F, Dejaco C, Matteson EL, Dasgupta B. Polymyalgia Rheumatica and Giant Cell Arteritis: A Systematic Review. JAMA. 2016;315(22):2442–58.
- Gozzoli DS, Hemmig A, Hemkens L, Werlen L, Ewald H, Berger C, et al. POS0806. Findings consistent with subclinical vasculitis in patients with new onset polymyalgia: a systematic literature review and a meta-analysis of cohort data. Ann Rheum Dis. 2021;80(Suppl 1):655–6.
- Gloor AD, Chollet L, Christ LA, Cullmann JL, Bonel HM, Villiger PM. Takayasu arteritis: Prevalence and clinical presentation in Switzerland. PLoS ONE. 2021;16(6):e0250025. 7. Onen F, Akkoc N. Epidemiology of Takayasu arteritis. Presse Med. 2017;46(7–8 Pt 2):e197–e203.
- Onen F, Akkoc N. Epidemiology of Takayasu arteritis. Presse Med. 2017;46(7–8 Pt 2):e197–e203.
- Aeschlimann FA, Barra L, Alsolaimani R, Benseler SM, Hebert D, Khalidi N, et al. Presentation and Disease Course of Childhood-Onset Versus Adult-Onset Takayasu Arteritis. Arthritis Rheumatol. 2019;71(2):315–23.
- Agard C, Barrier JH, Dupas B, Ponge T, Mahr A, Fradet G, et al. Aortic involvement in recent-onset giant cell (temporal) arteritis: a case-control prospective study using helical aortic computed tomodensitometric scan. Arthritis Rheum. 2008;59(5):670–6.
- Prieto-González S, Arguis P, García-Martínez A, Espígol-Frigolé G, Tavera-Bahillo I, Butjosa M, et al. Large vessel involvement in biopsy-proven giant cell arteritis: prospective study in 40 newly diagnosed patients using CT angiography. Ann Rheum Dis. 2012;71(7):1170–6.
- Gribbons KB, Ponte C, Carette S, Craven A, Cuthbertson D, Hoffman GS, et al. Patterns of Arterial Disease in Takayasu Arteritis and Giant Cell Arteritis. Arthritis Care Res (Hoboken). 2020;72(11):1615–24.
- Watanabe R, Berry GJ, Liang DH, Goronzy JJ, Weyand CM. Pathogenesis of Giant Cell Arteritis and Takayasu Arteritis-Similarities and Differences. Curr Rheumatol Rep. 2020;22(10):68.
- Weyand CM, Goronzy JJ. Immune mechanisms in medium and large-vessel vasculitis. Nature reviews Rheumatology. 2013;9(12):731–40.
- Ma-Krupa W, Jeon MS, Spoerl S, Tedder TF, Goronzy JJ, Weyand CM. Activation of arterial wall dendritic cells and breakdown of self-tolerance in giant cell arteritis. J Exp Med. 2004;199(2):173–83.
- Watanabe R, Maeda T, Zhang H, Berry GJ, Zeisbrich M, Brockett R, et al. MMP (Matrix Metalloprotease)-9-Producing Monocytes Enable T Cells to Invade the Vessel Wall and Cause Vasculitis. Circ Res. 2018;123(6):700–15.
- Piggott K, Biousse V, Newman NJ, Goronzy JJ, Weyand CM. Vascular damage in giant cell arteritis. Autoimmunity. 2009;42(7):596–604.
- Wu G, Mahajan N, Dhawan V. Acknowledged signatures of matrix metalloproteinases in Takayasu's arteritis. Biomed Res Int. 2014;2014:827105.
- Mahajan N, Dhawan V, Mahmood S, Malik S, Jain S. Extracellular matrix remodeling in Takayasu's arteritis: role of matrix metalloproteinases and adventitial inflammation. Arch Med Res. 2012;43(5):406–10.
- Seko Y, Minota S, Kawasaki A, Shinkai Y, Maeda K, Yagita H, et al. Perforin-secreting killer cell infiltration and expression of a 65-kD heat-shock protein in aortic tissue of patients with Takayasu's arteritis. J Clin Invest. 1994;93(2):750–8.
- Kurata A, Saito A, Hashimoto H, Fujita K, Ohno SI, Kamma H, et al. Difference in immunohistochemical characteristics between Takayasu arteritis and giant cell arteritis: It may be better to distinguish them in the same age. Mod Rheumatol. 2019;29(6):992–1001.
- Gloor AD, Berry GJ, Goronzy JJ, Weyand CM. Age as a risk factor in vasculitis. Semin Immunopathol. 2022.
- Wieczorek M, Abualrous ET, Sticht J, Álvaro-Benito M, Stolzenberg S, Noé F, et al. Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: Conformational Plasticity in Antigen Presentation. Front Immunol. 2017;8:292.
- Carmona FD, Coit P, Saruhan-Direskeneli G, Hernández-Rodríguez J, Cid MC, Solans R, et al. Analysis of the common genetic component of large-vessel vasculitides through a meta-Immunochip strategy. Sci Rep. 2017;7:43953.
- Deng J, Younge BR, Olshen RA, Goronzy JJ, Weyand CM. Th17 and Th1 T-cell responses in giant cell arteritis. Circulation. 2010;121(7):906–15.
- Saadoun D, Garrido M, Comarmond C, Desbois AC, Domont F, Savey L, et al. Th1 and Th17 cytokines drive inflammation in Takayasu arteritis. Arthritis Rheumatol. 2015;67(5):1353–60.
- Miyabe C, Miyabe Y, Strle K, Kim ND, Stone JH, Luster AD, et al. An expanded population of pathogenic regulatory T cells in giant cell arteritis is abrogated by IL-6 blockade therapy. Ann Rheum Dis. 2017;76(5):898–905.
- Jin K, Wen Z, Wu B, Zhang H, Qiu J, Wang Y, et al. NOTCH-induced rerouting of endosomal trafficking disables regulatory T cells in vasculitis. J Clin Invest. 2021;131(1).
- Maciejewski-Duval A, Comarmond C, Leroyer A, Zaidan M, Le Joncour A, Desbois AC, et al. mTOR pathway activation in large vessel vasculitis. J Autoimmun. 2018;94:99–109.
- Wen Z, Shen Y, Berry G, Shahram F, Li Y, Watanabe R, et al. The microvascular niche instructs T cells in large vessel vasculitis via the VEGF-Jagged1-Notch pathway. Sci Transl Med. 2017;9(399).
- Zhang H, Watanabe R, Berry GJ, Nadler SG, Goronzy JJ, Weyand CM. CD28 Signaling Controls Metabolic Fitness of Pathogenic T Cells in Medium and Large Vessel Vasculitis. J Am Coll Cardiol. 2019;73(14):1811–23.
- Hadjadj J, Canaud G, Mirault T, Samson M, Bruneval P, Régent A, et al. mTOR pathway is activated in endothelial cells from patients with Takayasu arteritis and is modulated by serum immunoglobulin G. Rheumatology (Oxford). 2018;57(6):1011–20.
- Borchers AT, Gershwin ME. Giant cell arteritis: a review of classification, pathophysiology, geoepidemiology and treatment. Autoimmun Rev. 2012;11(6–7):A544–54.
- González-Gay MA, García-Porrúa C, Llorca J, Hajeer AH, Brañas F, Dababneh A, et al. Visual manifestations of giant cell arteritis. Trends and clinical spectrum in 161 patients. Medicine (Baltimore). 2000;79(5):283–92.
- Kermani TA, Warrington KJ. Prognosis and monitoring of giant cell arteritis and associated complications. Expert Rev Clin Immunol. 2018;14(5):379–88.
- Maz M, Chung SA, Abril A, Langford CA, Gorelik M, Guyatt G, et al. 2021 American College of Rheumatology / Vasculitis Foundation Guideline for the Management of Giant Cell Arteritis and Takayasu Arteritis. Arthritis & rheumatology. 2021;73(8):1349–65.
- Monti S, Bartoletti A, Bellis E, Delvino P, Montecucco C. Fast-Track Ultrasound Clinic for the Diagnosis of Giant Cell Arteritis Changes the Prognosis of the Disease but Not the Risk of Future Relapse. Front Med (Lausanne). 2020;7:589794.
- Hellmich B, Agueda A, Monti S, Buttgereit F, de Boysson H, Brouwer E, et al. 2018 Update of the EULAR recommendations for the management of large vessel vasculitis. Annals of the rheumatic diseases. 2020;79(1):19–30.
- Schirmer JH, Aries PM, Balzer K, Berlit P, Bley TA, Buttgereit F, et al. S2k-Leitlinie: Management der Großgefässvaskulitiden. Zeitschrift für Rheumatologie. 2020;79(3):67–95.
- Stahn C, Buttgereit F. Genomic and nongenomic effects of glucocorticoids. Nat Clin Pract Rheumatol. 2008;4(10):525–33.
- Villiger PM, Adler S, Kuchen S, Wermelinger F, Dan D, Fiege V, et al. Tocilizumab for induction and maintenance of remission in giant cell arteritis: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet. 2016;387(10031):1921–7.
- Stone JH, Tuckwell K, Dimonaco S, Klearman M, Aringer M, Blockmans D, et al. Trial of Tocilizumab in Giant-Cell Arteritis. N Engl J Med. 2017;377(4):317–28.
- Christ L, Seitz L, Scholz G, Sarbu A-C, Amsler J, Bütikofer L, et al. Tocilizumab monotherapy after ultra-short glucocorticoid administration in giant cell arteritis: a single-arm, open-label, proof-of-concept study. Lancet Rheumatol. 2021;3(9):e619-e26.
- Mahr AD, Jover JA, Spiera RF, Hernandez-Garcia C, Fernandez-Gutierrez B, Lavalley MP, et al. Adjunctive methotrexate for treatment of giant cell arteritis: an individual patient data meta-analysis. Arthritis Rheum. 2007;56(8):2789–97.
- Hellmich B, Agueda AF, Monti S, Luqmani R. Treatment of Giant Cell Arteritis and Takayasu Arteritis-Current and Future. Current rheumatology reports. 2020;22(12):84.



